A major challenge facing researchers today is the cleanup and harmonization of data for use in their work. As datasets get larger and the types of outputs continue to grow, this problem will continue to grow exponentially. Manually cleaning this data can be time-consuming and prone to human error.
Addressing these needs for cleanup and harmonization of functional genomics data is how hipFG (Harmonization and Integration Pipeline for Functional Genomics), a high-throughput harmonization and integration pipeline, was born. We sat down with Jeffrey Cifello, lead code developer for the hipFG project, to learn about hipFG and how it can help researchers in their work.
What prompted the development of hipFG?
Well, we have another resource in the lab, called FILER (Functional Genomics Repository), which is a set of large, curated, and harmonized functional genomics and annotation data which can be searched to check researchers results against what is already out there.
We wanted to add new datasets to expand this resource but were running into a lot of difficult ad hoc nuances for each new data set. It was taking a lot of time to manually to go in, identify where our scripts needed to be modified, visually check them, run them, debug them, all complicated by the many places where human error could be introduced. So, we set about trying to automate this process.
First, we started by looking at what was already available, and there wasn’t a ton for public use for functional genomics. There is the eQTL catalogue, but their pipeline is not open source to our knowledge and is very specific to the type of work they do. There is also MungeSumStats, but that is only for TSV/VCF files and, while it can handle summary statistics, it doesn’t allow for secondary target QTLs (quantitative trait loci), or, say Hi-C chromatin data.
So clearly there was an unmet need in the space for a tool that could harmonize data, how did you go about creating hipFG?
Definitely. We worked together as a team. Dr. Pavel Kuksa led the conceptualization, like what does it mean for an allele to be verified? What do we want out outputs to look like? What is the user experience going to be like? I handled all the coding and from that process determined what our inputs should look like. Dr. Fanny Leung helped us clarify the types of inputs and outputs we were interested in. Otto Valladares supported us from an IT development perspective and Naveensri Saravanan was our beta tester, making sure everything was downloadable, ran, and helped me develop the user documentation. All in all, it took about a year to get to a mature workable state.
And how does hipFG work?
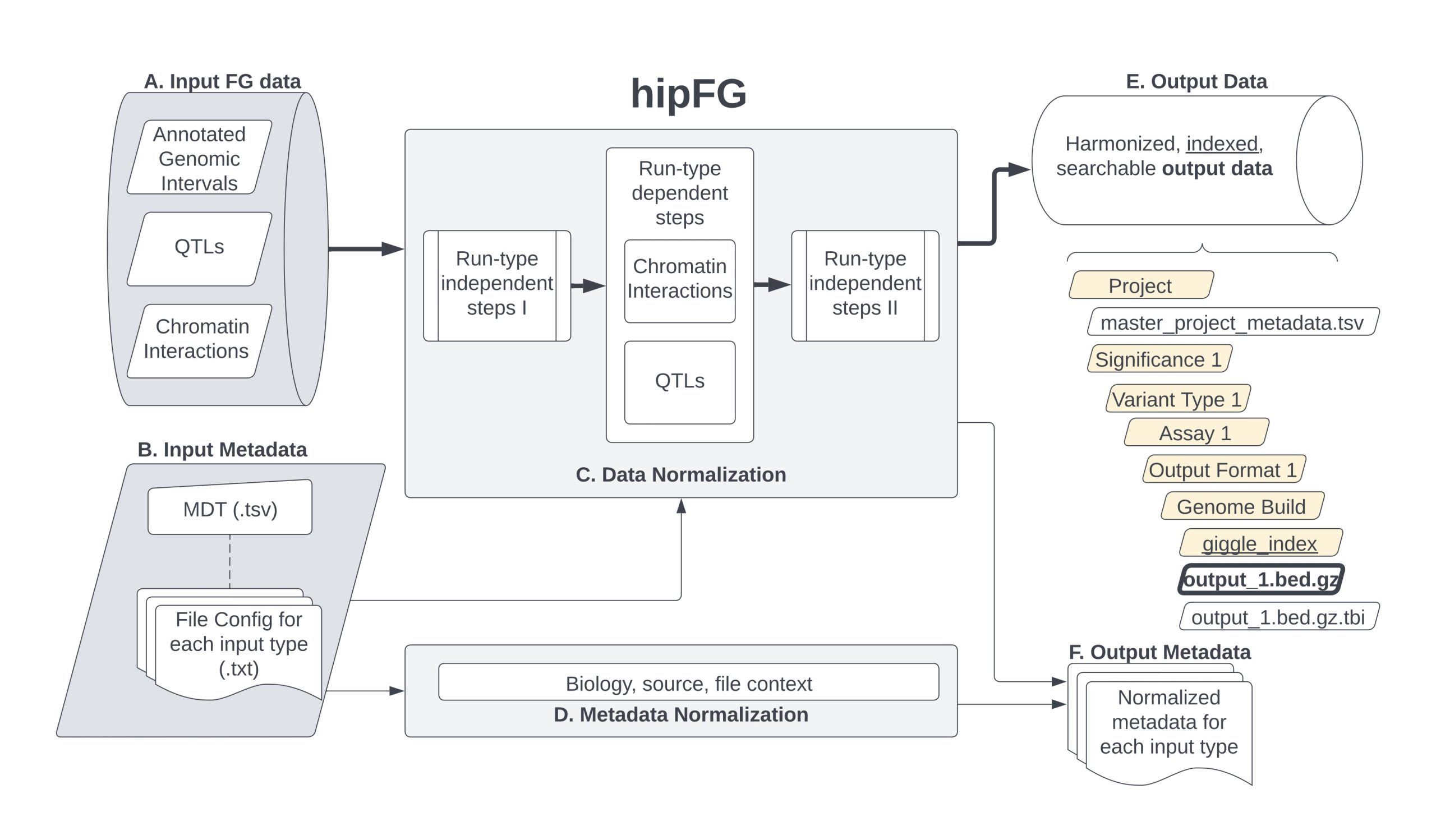
hipFG sits on Python and Bash scripts. The user points out where their input is and maps their input columns to the output columns corresponding to the input data type. The input types to choose from are (1) Genomics annotations, (2) QTLs, and (3) Chromatin interactions. hipFG then automatically picks from a pool of scripts to create a custom pipeline script for the input data. hipFG’s output is genomic annotations, but we have an output format developed for compatibility with bedtools, Giggle, and building FILER. The bottom-line is that hipFG can have any number of input formats, as long as it’s a TSV pointing to genomic positions.
It can be run in command line or through Jupyter notebooks we put together. We recommend first time users look at the example docs and modify those to fit their needs.
That sounds like a great tool, how can researchers apply this to their work?
The goal of hipFG is really to make functional genomics data more accessible for analysis by simplifying and streaming those clean up steps.
Researchers can use it to clean their own data before sharing to ensure that everyone is starting from the same point when trying to replicate work or examine the same dataset a different way. Or researchers can use it to clean data they have gotten from others, which can be especially helpful if the data is coming from multiple sources.
It helps researchers save time by reducing human error, preventing false results from small code mistakes.
It also puts everything into a systematic hierarchy, stores outputs in the same place in the same way, meaning you won’t likely misplace a dataset, saving time and instilling confidence in your data.
What type of resources are there to help people learn about hipFG?
In our documentation, it works through examples for how to run the three major data types, the input files and expected outcome.
There is a readme that will do a test run and will show if the test input corresponds to the test output so you can see if things are running correctly.
There are also a handful of tutorials specific to input, configuration, and an advanced usage guide, which has various additional features we didn’t have space to mention in the paper but have found to be super helpful in our own work like: splitting info fields on the fly, splitting QTLs, splitting associations from nominal, false discovery, and all associations.
And then we also have the Jupyter notebooks that users can take and adapt to their own needs.
What impact do you hope hipFG will have for the community?
I hope that hipFG will allow researchers to have more time for analysis and asking interesting questions by letting them spend less time cleaning data!
Check out the hipFG paper to learn more about the development process and how to apply it to your research.