By Fernanda Ruiz, NOVA Research Company
Alzheimer’s disease (AD) is a degenerative disease that likely is caused by a combination of environmental, lifestyle, and genetic factors, and increasing age. Alzheimer’s Disease Sequencing Project (ADSP) researchers are focused on gaining further insights into the genetic component of this disease, including the role of structural variants (SVs), which could lead to identification of novel therapeutic targets.
SVs are defined generally as genomic variations larger than 50 base pairs (bp) and can be very diverse. The main types of SVs are deletions, duplications, inversions, insertions, and translocations. Copy number variation (CNV) is a subtype of SV that is generated by duplications and deletions; this results in a variable copy number of a DNA region.
Multiple studies have shown that SVs are linked to AD. For example, CNVs in known AD risk genes (e.g., CR1) have been identified as well as CNVs that are known to cause Mendelian early-onset AD. Despite the many studies exploring genetic variations in AD, research is needed to further discover AD-associated SVs and their contribution to the disease.
Dr. Wan-Ping Lee, a Research Assistant Professor of Pathology and Laboratory Medicine at the University of Pennsylvania, oversees information technology (IT)-related projects at the Penn Neurodegeneration Genomics Center (PNGC) and performs SV and CNV detection on ADSP whole-genome sequencing (WGS) data (see the SV Discovery Pipeline below).
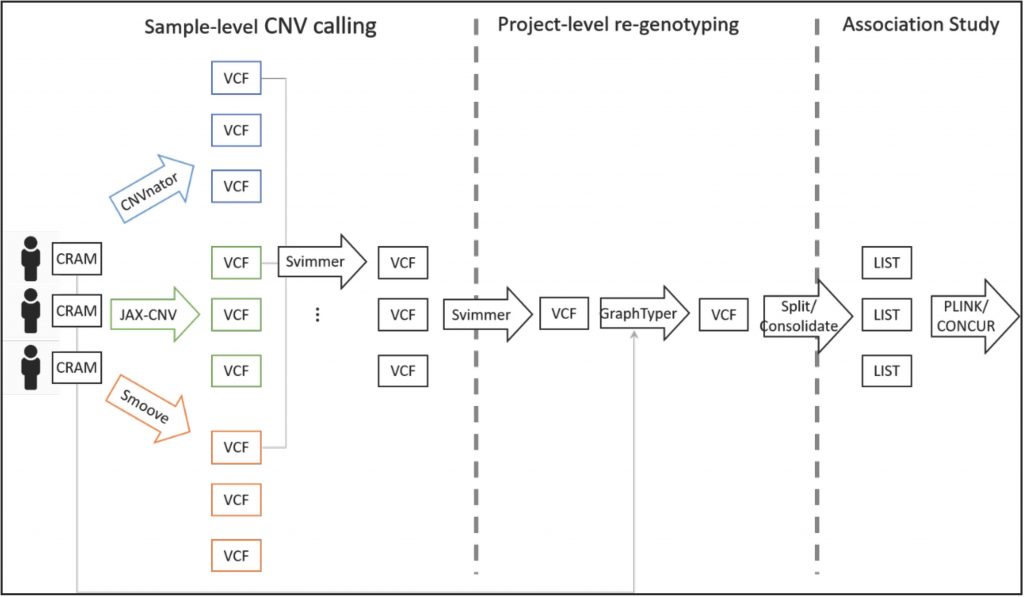
According to Dr. Lee, identification of SVs and CNVs is challenging because both size and position must be taken into account. SV complexity (e.g., overlapping or nesting) further complicates their detection.
ADSP investigators are meeting this challenge by utilizing novel and efficient computational methods that leverage next-generation sequencing data (i.e., WGS). These tools use different approaches—detection of changes in read depth, identification of read pairs or split reads, or de novo genome assembly. SV validation can be accomplished by comparing the call set with other call sets from large WGS projects (e.g., the 1000 Genomes Project and the Genome Aggregation Database [gnomAD]), using array data, or conducting experimental validation.
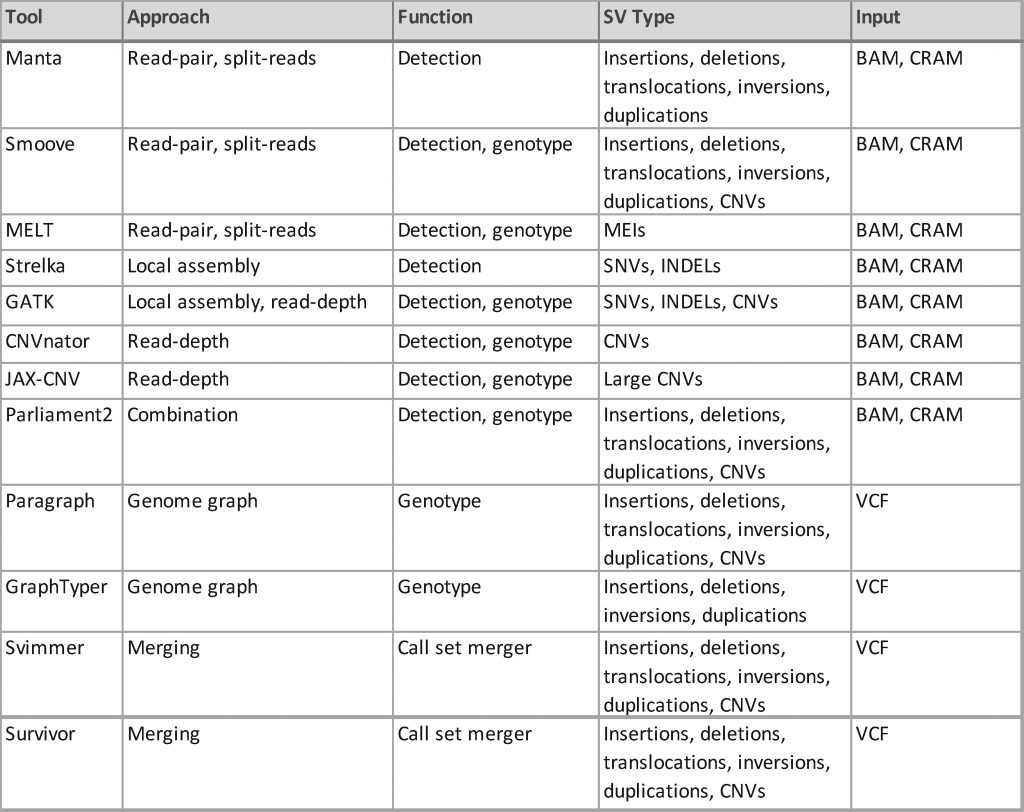
Investigators, including those at PNGC, are using a variety of tools to discover and genotype SVs relevant to AD from short-read sequencing data that vary by function (i.e., detection, genotyping, merging), SV types and sizes detected, approach, and input type such as binary alignment map (BAM), compressed BAM (CRAM), or variant call format (VCF).
It should be noted that studies evaluating SV tools use different assessment methods, and no single study has compared them all; thus, it is difficult to identify the superior tool. Details about these tools are provided below (also see the Overview of SV Tools table).
- Manta is a rapid, general-purpose caller that detects insertions, deletions, translocations, inversions, and duplications. Manta uses both read-pairs and split-read approaches and can be applied to one sample or a small set of samples. Manta accepts CRAM and BAM files and is publicly available.
- Smoove can detect and genotype insertions, deletions, translocations, inversions, duplications, and CNVs rapidly using both read-pairs and split-read approaches. This tool is publicly available and can be applied to small and large cohorts. The specificity of this tool is improved due to exclusion of spurious alignment signals. The input can be CRAM or BAM files.
- Mobile Element Locator Tool (MELT) identifies, annotates, and genotypes mobile element insertions (MEIs) in large WGS datasets using both read-pairs and split-read approaches. Developed as part of the 1000 Genomes Project for identification of MEIs at a population level, this publicly available tool is faster and has higher sensitivity and specificity than other MEI-specific tools. It is also scalable and can be adapted for use with parallel platforms. The input can be CRAM or BAM files.
- Strelka uses a local assembly approach to detect single nucleotide variants (SNVs) and short insertions and deletions (INDELs smaller than 50 bp) for paired tumor-normal samples. This method is optimized for a small number of samples but also works for case/control or population-level sample sets. This tool is publicly available. Input can be BAM or CRAM files.
- Genome Analysis Toolkit (GATK) is a highly accurate tool that is used routinely to discover and genotype SNVs and INDELs. A new feature of CNV identification recently was released. This tool uses a read-depth-based approach and is publicly available. The input can be CRAM or BAM files.
- CNVnator can detect CNVs (including atypical CNVs) using a read-depth-based approach with high sensitivity and specificity. CNVnator can identify large CNVs precisely and is publicly available. This tool also can genotype SVs and accepts CRAM and BAM files. This caller cannot detect CNVs generated by transposable elements.
- JAX-CNV detects large (>50 kb) CNVs with high sensitivity and a very low false discovery rate using a read-depth approach. JAX-CNV is superior to other callers in identifying known disease-related duplications and deletions. Computation time required to analyze one sample is shorter compared with other tools. It is also easy to use. The input can be CRAM or BAM files.
- Parliament2 combines tools (Breakdancer, Breakseq2, Delly2, Manta, and Lumpy) for discovery of deletions, insertions, duplications, inversions, and translocations rapidly and with high accuracy. This tool also can genotype SV candidates. Running different callers in parallel reduces the time and cost for SV discovery in many samples (thousands); it also can be applied to a single sample. Parliament2 is publicly available. The input can be CRAM or BAM files. One limitation is that Parliament2 cannot identify SVs smaller than 50bp.
- Paragraph accurately genotypes previously identified SVs using sequence graphs (i.e., genome graphs) and SV annotations. Paragraph can rapidly genotype variants identified using different SV callers and is scalable. This tool is publicly available and accepts VCF files. One disadvantage is that only one SV per locus can be genotyped.
- GraphTyper rapidly and accurately genotypes deletions, insertions, duplications, and inversions by realigning short-read sequence data to a pangenome. This tool can be applied to small and large datasets and is more sensitive and scalable compared with other SV callers. The input is a VCF file. Limitations include reference bias and ambiguous alignments.
- SV merging tools like Svimmer and Survivor group same-type SVs to produce a single VCF file containing the merged SV sites. The regions in these VCFs then can be genotyped using tools like GraphTyper.
In addition to the capabilities described above, key considerations for selection of SV tools include computational burden/cost as well as tool sensitivity, specificity, and false discovery rates. The variability of SV tool functions also hinders researchers from selecting the best tool to answer their specific scientific questions. According to Dr. Lee, “An ideal SV caller would handle all types of SVs across the full spectrum of SV lengths,” but such a tool does not exist.
Dr. Lee is enthusiastic about searching for impactful SVs and CNVs that will shed light into AD mechanisms and therapeutic targets. She and other ADSP investigators will continue to utilize a wide range of SV tools to investigate novel SVs linked to AD.
Learn more about Dr. Lee on her Penn faculty page and about PNGC scientific projects here.
This is a collaborative research project between Penn, Boston University, Case Western Reserve University, Columbia University, and the University of Miami. Special thank to Drs. Gerard D. Schellenberg, Li-San Wang, Fanny Leung, John Malamon, John Farrell, Lindsay Farrer, Badri Vardarajan, Jonathon Haines, Will Bush, Anita DeStefano, and Kim Worley, as well as the valued team members, Amanda Kuzma, Yi-Fan Chou, and Otto Valladares.